
Leveraging genome annotations and shared genetic factors across multiple diseases,
we develop computationally efficient and statistically powerful methods to identify
high risk individuals for better disease monitoring and prevention.
We also investigate how non-genetic factors interact with genetic susceptibility to impact disease risks.
[1] L. Xu, W. Zheng, J. Hu, Y. Lin, J. Zhao, G. Wang, T. Liu, H. Zhao (2025) Improving polygenic risk prediction performance through integrating electronic health records by phenotype embedding. American Journal of Human Genetics, 112: 3030-3045.
[2] Zhou G, Yolou I, Xie Y, Zhao H (2025) Leveraging local ancestry and cross-ancestry genetic architecture to improve genetic prediction of complex traits in admixed populations. American Journal of Human Genetics, 112: 1923-1935.
[3] Xu L, Zhou G, Jiang W, Zhang H, Dong Y, Guan L, Zhao H (2025) JointPRS: A data-adaptive framework for multi-population genetic risk prediction incorporating genetic correlation. Nature Communications, 16: 3841.
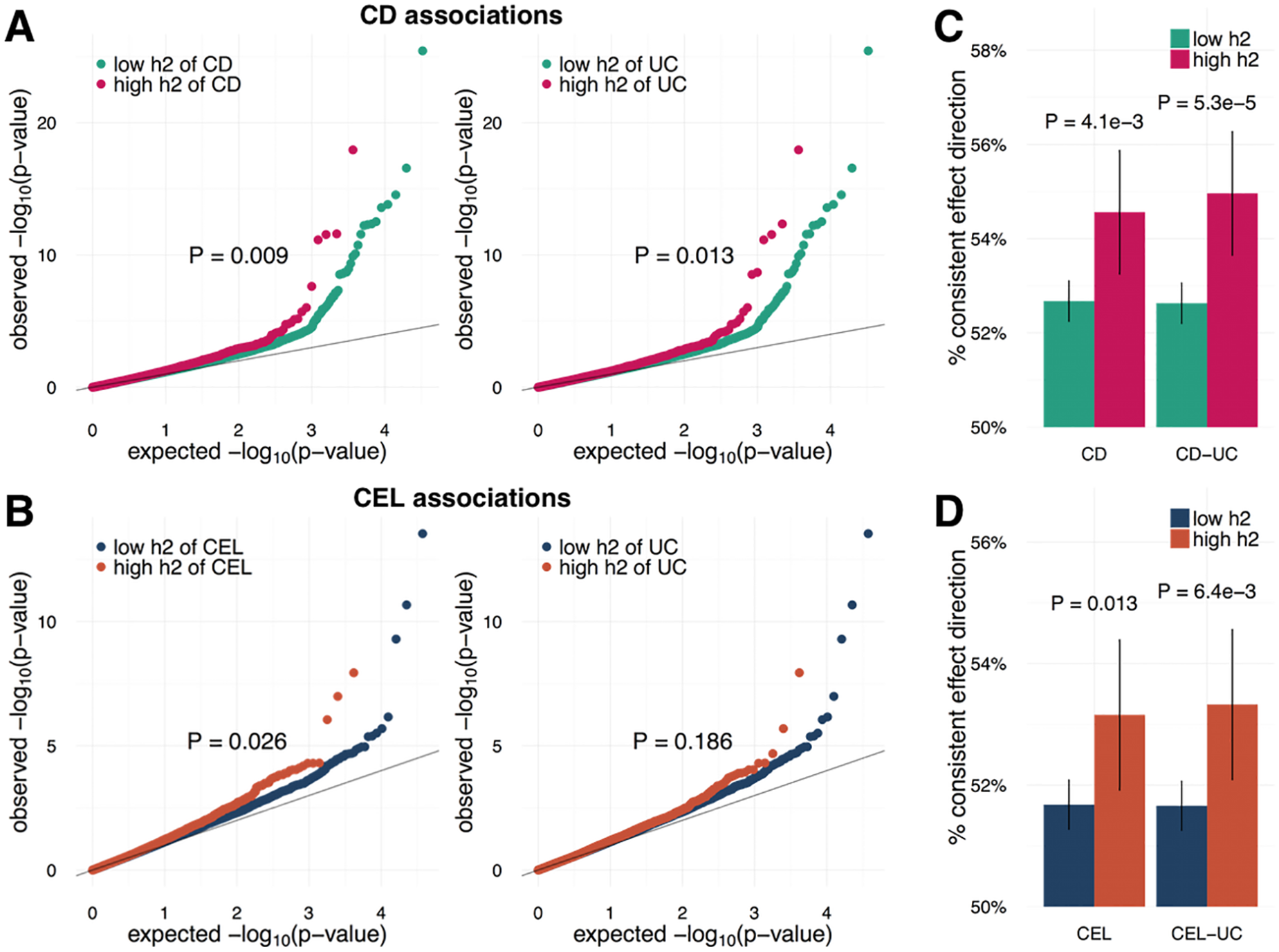
[4] Hu J, Ye Y, Zhang C, Ruan Y, Natarajan P, Zhao H (2025) Robust pleiotropy-decomposed polygenic scores identify distinct contributions to elevated coronary artery disease polygenic risk. PLOS Computational Biology, 21: e1013191.
[5] G. Zhou, X. Qie, H. Zhao (2025) A Bayesian approach to correcting the attenuation bias of regression using polygenic risk score. Genetics, 4: iyaf018.
[6] M. T. Sheikh, H. Zhao (2025) A semicompeting risks model with an application to UK Biobank data to identify risk factors for diabetes onset and progression. Biometrics, 81: ujaf003.
[7] W. Jiang, L. Chen, M. J. Girgenti, H. Zhao (2024) Tuning parameters for polygenic risk score methods using GWAS summary statistics from training data. Nature Communications, 15: 24.
[8] G. Zhou, T. Chen, H. Zhao (2023) SDPRX: A statistical method for cross-population prediction of complex traits. The American Journal of Human Genetics, 110:1.
[9] G. Zhou, H. Zhao (2021) A fast and robust Bayesian nonparametric method for prediction of complex traits using summary statistics. PLOS Genetics, 17: e1009697.
[10] S. Song, W. Jiang, L. Hou, H. Zhao (2020) Leveraging effect size distributions to improve polygenic risk scores derived from summary statistics of genome-wide association studies. PLOS Computational Biology, 16: e1007565.
[11] Y. Hu, Q. Lu, W. Liu, Y. Zhang, M. Li, H. Zhao (2017) Joint modeling of genetically correlated diseases and functional annotations increases accuracy of polygenic risk prediction. PLOS Genetics, 13: e1006836.
[12] Y. Hu, Q. Lu, R. Powles, X. Yao, C. Yang, F. Fang, X. Xu, H. Zhao (2017) Leveraging functional annotations in genetic risk prediction for human complex diseases. PLOS Computational Biology, 13: e1005589.
[13] C. Li, C. Yang, J. Gelernter, H. Zhao (2014) Improving genetic risk prediction by leveraging pleiotropy. Human Genetics, 133: 639-650.
© 2022 Hongyu Zhao, Ph.D.
Created by Eddie, Chen and Wangjie.