
We develop statistical methods to annotate the human genome using diverse data sources (e.g. sequence, ENCODE, Roadmap Epigenomics, GTEx)
and apply these annotations to better analyze and interpret the association signals identified from numerous genome wide association studies.
We also develop methods to integrate diverse data types and results from different phenotypes to better identify functional genes/variants,
as well as methods to characterize genetic architecture of complex traits.
[1] C. Lin, Y. Lin, W. Li, L. Xu, X. Zhang, H. Zhao. Leveraging cell-type specificity and similarity improves single-cell eQTL fine-mapping. Nature Communications, in press.
[2] X. Zhang, L. Wang, J. Zhao, H. Zhao (2025) Knockoff procedure improves causal gene identifications in conditional transcriptome-wide association studies. American Journal of Human Genetics, 112: 2476-2492.
[3] Z. Dong, W. Jiang, J. Shen, H. Li, Y. Xie, A. T. DeWan, H. Zhao (2025) Incorporating additive genetic effects and linkage disequilibrium information to discover gene-environment interactions using BV-LDER-GE. Genome Biology, 26: 332.
[4] Cheng Y, Zhou G, Li H, Zhang X, Justice A, Martinez C, Aouizerat BE, Xu K, Zhao H (2025) Incorporating local ancestry information to predict genetically associated DNA methylation in admixed populations. Briefings in Bioinformatics, 26: bbaf325.
[5] Z. Dong, W. Jiang, H. Li, A. T. DeWan, H. Zhao (2024) LDER-GE estimates phenotypic variance component of gene-environment interactions in human complex traits accurately with GE interaction summary statistics and full LD information. Briefings in Bioinformatics, 25: bbae335.
[6] X. Zhang, W. Jiang, H. Zhao (2024) Integration of expression QTLs with fine mapping via SuSiE. PLOS Genetics, 20: e1010929.
[7] Liu W, Deng W, Chen M, Dong Z, Zhu B, Yu Z, Tang D, Sauler M, Lin C, Wain LV, Cho MH, Kaminski N, Zhao H (2023) A statistical framework to identify cell types whose genetically regulated proportions are associated with complex diseases. PLOS Genetics, 19: e1010825.
[8] J. Jiang, W. Jiang, D. Paul, Y. Zhang, H. Zhao (2023) High dimensional asymptotic behavior of inference based on GWAS summary statistics. Statistica Sinica, in press.
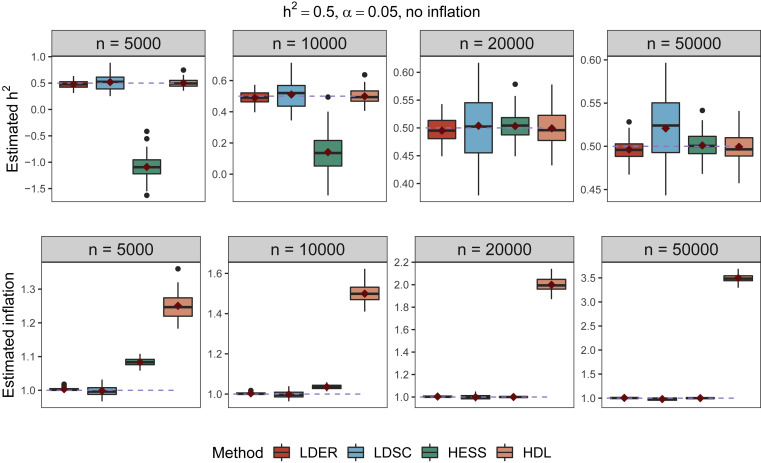
[9] S. Song, W. Jiang, Y. Zhang, L. Hou, H. Zhao (2022) Leveraging LD eigenvalue regression to improve the estimation of SNP heritability and confounding inflations. American Journal of Human Genetics, 109: 802-811.
[10] X. Hu, J. Zhao, Z. Lin, Y. Wang, H. Peng, H. Zhao, X. Wan, C. Yang (2022) Mendelian Randomization for causal inference accounting for pleiotropy and sample structure using genome-wide summary statistics. PNAS, 119: e2106858119.
[11] Y. Zhang, Q. Lu, Y. Ye, K. Huang, W. Liu, Y. Wu, X. Zhong, B. Li, Z. Yu, B. Travers, D. Werling, J. Li, H. Zhao (2021) SUPERGNOVA: Local genetic correlation analysis reveals heterogeneous etiologic sharing of complex traits. Genome Biology, 22: 262.
[12] W. Liu, M. Li, W. Zhang, G. Zhou, X. Wu, J. Wang, Q. Lu, H. Zhao (2020) Leveraging functional annotation to identify genes associated with complex diseases. PLOS Computational Biology, 16: e1008315.
[13] Y. Hu, M. Li, Q. Lu, H. Weng, J. Wang, S. M. Zekavat, Z. Yu, B. Li, J. Gu, S. Muchnik, Y. Shi, B. W. Kunkle, S. Mukherjee, P. Natarajan, A. Naj, A. Kuzma, Y. Zhao, P. K. Crane, Alzheimer's Disease Genetics Consortium, H. Lu, H. Zhao (2019) A statistical framework for cross-tissue transcriptome-wide association analysis. Nature Genetics, 51: 568-576.
[14] Q. Lu, B. Li, D. Ou, M. Erlendsdottir, R. L. Powles, T. Jiang, Y. Hu, D. Chang, C. Jin, W. Dai, Q. He, Z. Liu, S. Mukherjee, P. K. Crane, H. Zhao (2017) A powerful approach to estimating annotation-stratified genetic covariance using GWAS summary statistics. American Journal of Human Genetics, 101: 939-964.
[15] Q. Lu, R. L. Powles, S. Abdallah, D. Ou, Q. Wang, Y. Hu, Y. Lu, W. Liu, B. Li, S. Mukherjee, P. K. Crane, H. Zhao (2017) Systematic tissue-specific functional annotation of the human genome highlights immune-related DNA elements for late-onset Alzheimer’s disease. PLOS Genetics, 13: e1006933.
[16] Q. Lu, R. Powles, Q. Wang, J. He, H. Zhao (2016) Integrative tissue-specific functional annotations in the human genome provide novel insights on many complex traits and improve signal prioritization in genome wide association studies. PLOS Genetics, 12: e1005947.
[17] J. Jiang, C. Li, D. Paul, C. Yang, H. Zhao (2016) On high-dimensional misspecified mixed model analysis in genome-wide association study. Annals of Statistics, 44: 2127–2160.
[18] D. Chung, C. Yang, C. Li, J. Gelernter, H. Zhao (2014) GPA: A statistical approach to prioritizing GWAS results by integrating pleiotropy and annotation. PLOS Genetics, 10: e1004787.
[19] L. Hou, M. Chen, C. K. Zhang, J. Cho, H. Zhao (2014) Guilt by Rewiring: Gene prioritization through network rewiring in genome wide association studies. Human Molecular Genetics, 23: 2780-2790.
[20] M. Chen, J. Cho, H. Zhao (2011) Incorporating biological pathways via a Markov random field model in genome-wide association studies. PLoS Genetics, 7: e1001353.
© 2022 Hongyu Zhao, Ph.D.
Created by Eddie, Chen and Wangjie.